Software Description
Algorithm summary:
After being called, SNAPPY searches for a .raw file with the correct prefix (controlled by parameter ‘infile’). If the .raw file is not available, SNAPPY calls plink to create the .raw file from the plink library determined by the ‘infile’ parameter. SNAPPY then creates a set of reference dictionaries to track the relationships between various SNP identifiers, positions, ancestral and derived alleles, and associated haplogroups. These reference dictionaries are built from the set of reference files that are included in the program distribution.
Genotypes from all samples, read in from the .raw file, are then stored as a list of dictionaries, with each dictionary containing key-value pairs consisting of Y-chromosome positions (keys) and allele (values) for each sample. SNAPPY then cycles through each sample’s genotypes and each Y-chromosome haplogroup stored in the reference dictionaries, counting the number of haplogroup-informative alleles present in the sample. This number, when divided by the sample’s number of non-missing haplogroup-informative positions, is the haplogroup’s score for the given sample. To illustrate, consider a haplogroup that is defined by 5 SNPs, and an individual who has been genotyped at these 5 sites. If the individual is missing one genotype, and has the derived allele for three of the sites, and the ancestral allele at the fifth site, then the score is 3/4=0.75. Importantly, a particular haplogroup’s score uses alleles from both its own haplogroup-informative positions as well as all its ancestral haplogroups (Figure 1). If all informative positions are missing for a given haplogroup, the score for the haplogroup is set to zero. Additionally, if no informative alleles from a particular haplogroup’s two most recent ancestors are present, that haplogroup will not be considered for assignment to a sample (e.g., referenced node in Figure 1C would not be considered because its two most recent ancestors lack informative alleles in the sample). Each haplogroup is evaluated independently for every individual, and the scores are stored in a two-dimensional numpy array to allow for efficient storage and quick processing.
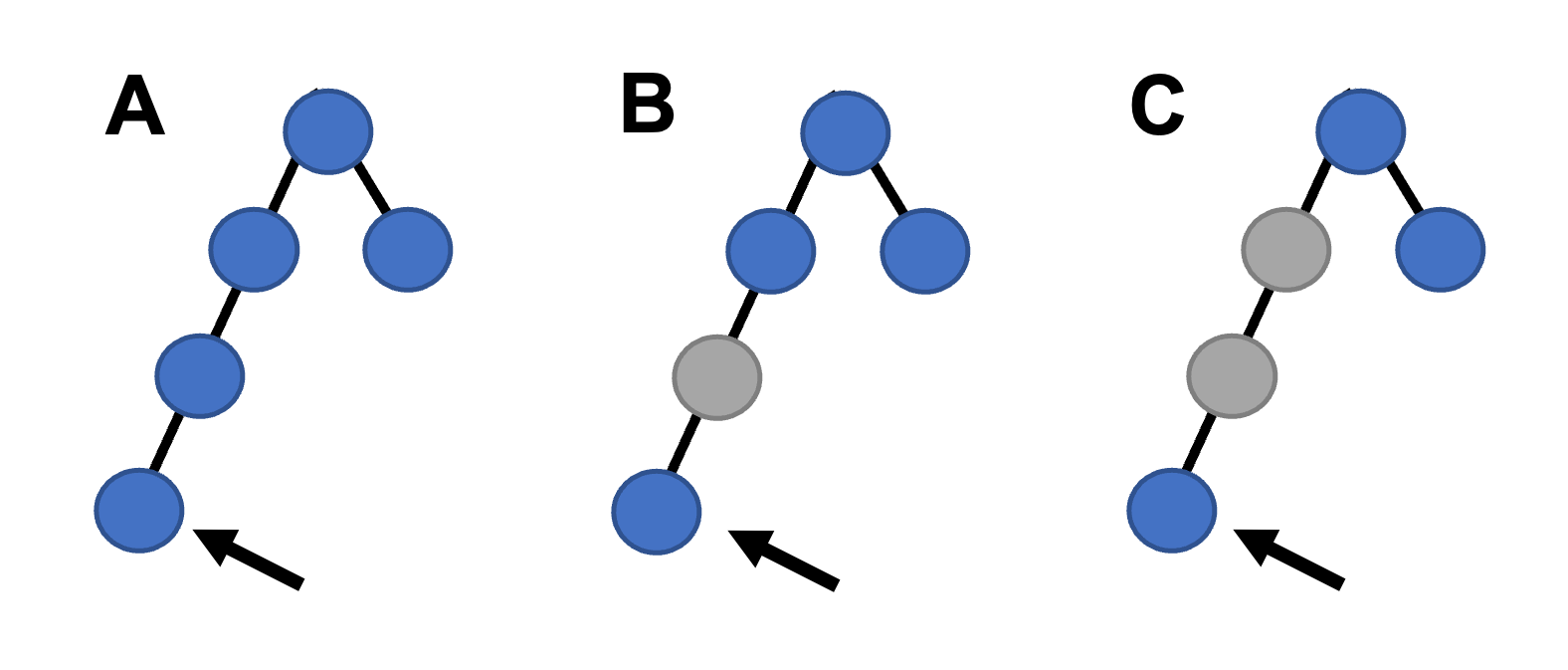
Figure 1 - Possible ancestral haplogroup patterns used to inform if the indicated haplogroup (arrow) is supported by genotype data. Blue circles indicate the presence of haplogroup-informative alleles for a haplogroup, while gray represents haplogroups for which no informative alleles are present.
SNAPPY then makes haplogroup assignments to the deepest node with sufficient support by first considering all haplogroup nodes that have a score greater than a user-defined threshold (default=0.6, controlled by parameter ‘min_hap_score’) and no descendant haplogroups with scores greater than the threshold. SNAPPY makes the haplogroup assignment based on the haplogroup with the highest score, or may make the assignment to the deepest haplogroup with a score higher than a user-defined threshold (default=0.8, controlled by parameter ‘min_deep_score’), depending on the value of the min_hap_score and min_deep_score parameters. The values of both of these parameters can be adjusted at the command line at runtime if the user wishes to prioritize deeper haplogroup assignments vs. higher-scoring assignments.
Required input files:
For convenient use, SNAPPY accepts input data formatted as a common plink binary library consisting of a .bed file, a .bim file, and a .fam file, each with the same base name, or as a .vcf file. Positions on autosomes, the mitochondrial genome, or the X-chromosome should be filtered out prior to running SNAPPY. Other necessary input files that are used to read and store SNP-haplogroup assignments, and haplogroup ancestor-descendant relationships on the Y-chromosome tree are included in the SNAPPY distribution in the ‘ref_files’ directory.
Output files:
After performing assignments, SNAPPY writes two output files. The first, the .out file (default= chrY_hgs.out, but controlled by the ‘out’ parameter), is a tab-separated file where each line gives a sample id, the sample’s haplogroup assignment, the haplogroup score, and the list of that haplogroup’s informative alleles used in determining the score. The second file, the .all file (default=chrY_hgs.all, but controlled by the ‘out’ parameter), is a tab-separated file where each line lists the sample number followed by every haplogroup that exceeded a threshold score (see Parameters section) in the format ‘Hapologroup:Score.’ This allows users to manually adjust haplogroup assignments where necessary.
Reference File Sources:
Files included in the ‘ref_files’ directory include: pos_to_allele.txt, id_to_pos.txt, y_hg_and_snps.sort, and tree_structure.txt. The first three files contain information about positions and id’s of snps on the Y-chromosome, and on to which haplogroups are informed by the snps. The final file, tree_structure.txt, details information on haplogroup descent where parent or child haplogroup names do not follow the Y-chromosome haplogroup naming conventions. These files were created from Y-chromosome trees maintained by the International Society of Genetic Genealogy (ISOGG), and from discussions with experts in Y-chromosome history.
We anticipate updating reference files periodically and will make them available to the public in the SNAPPY GitHub repository. In addition, users may easily create their own reference files and haplogroup databases by following the format of each of these files. Note that tree_structure.txt is formatted as “parent haplogroup-TAB-child haplogroup.” Please also note that custom Y-chromosome libraries must follow the exact names of the provided reference files unless specified with an optional argument.
Dependencies:
SNAPPY is implemented in python2 (SNAPPY_v0.2.1) and in python3 (SNAPPY_v0.2.2) and makes use of the python modules ‘numpy’, ‘sys’, ‘os’, ‘os.path’, ‘re’, and ‘subprocess’. In addition, a plink (v1.9) executable must be listed in the user’s path as ‘plink’ for preprocessing steps. plink is available for all major operating systems and can be downloaded here.